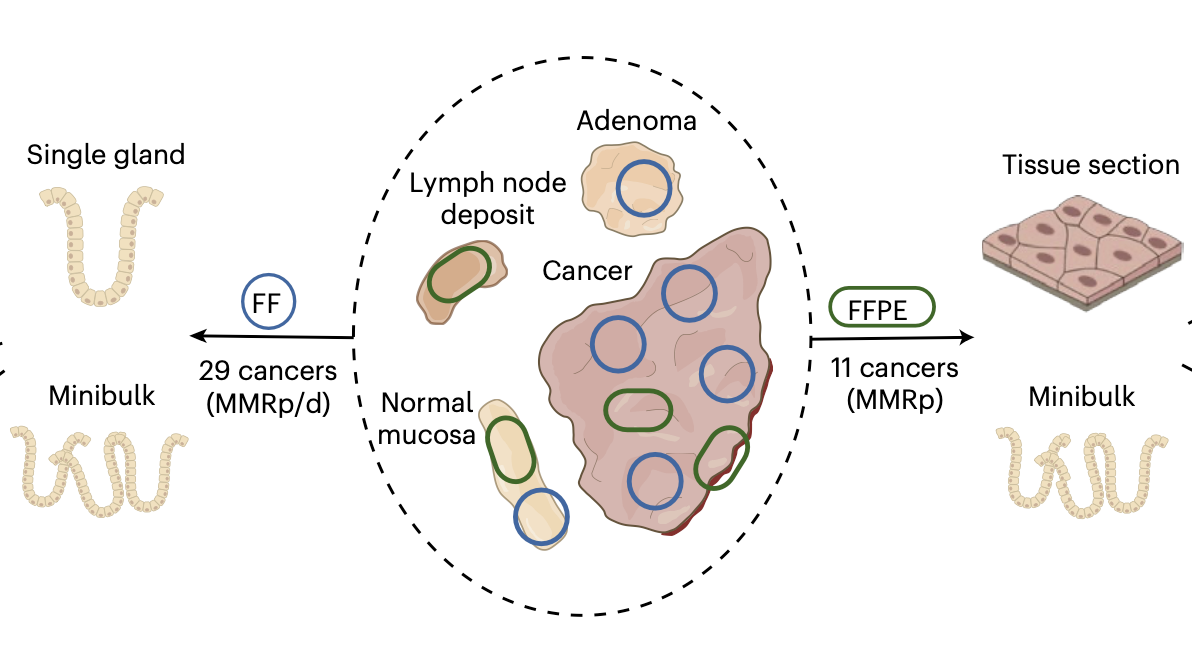
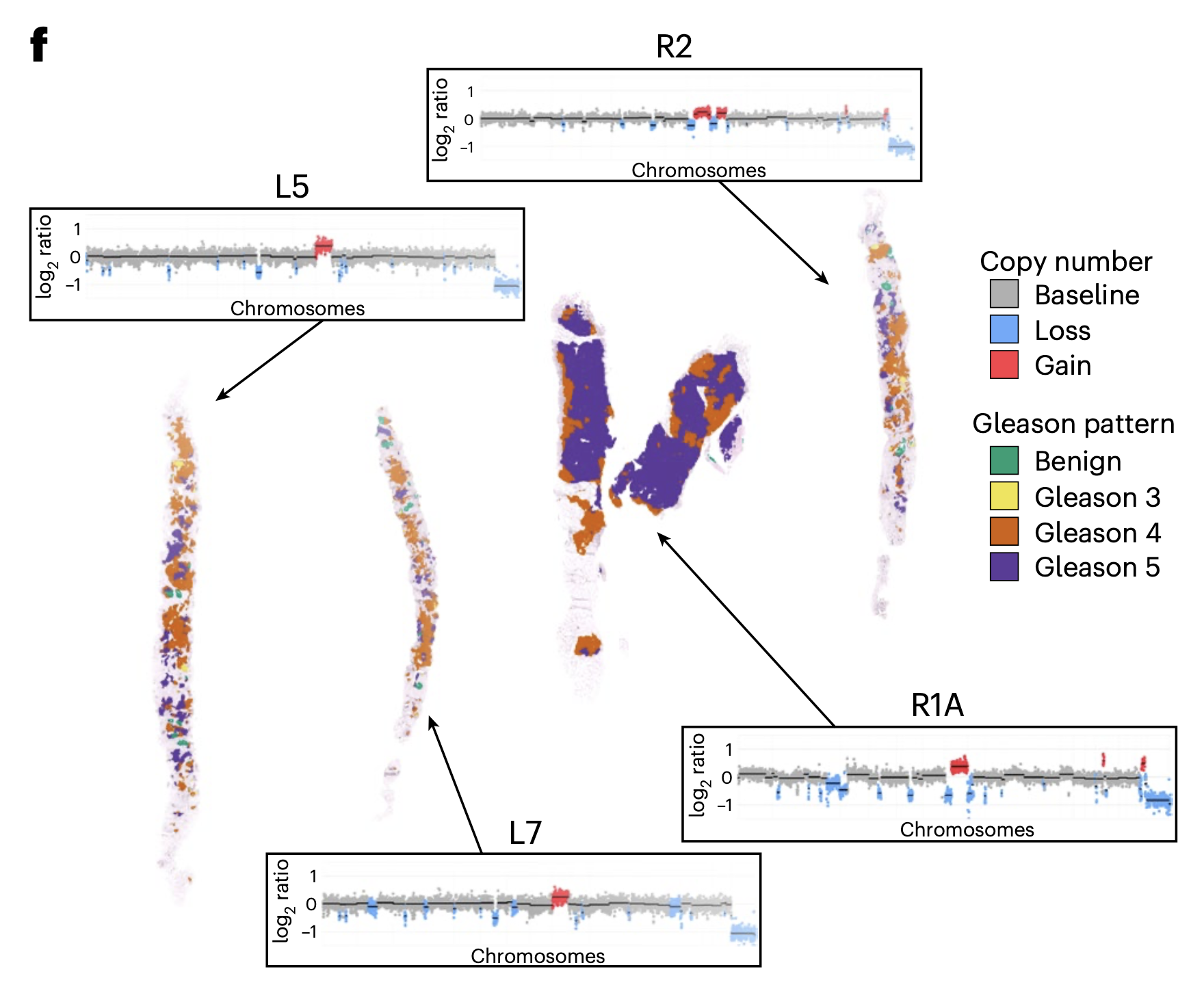
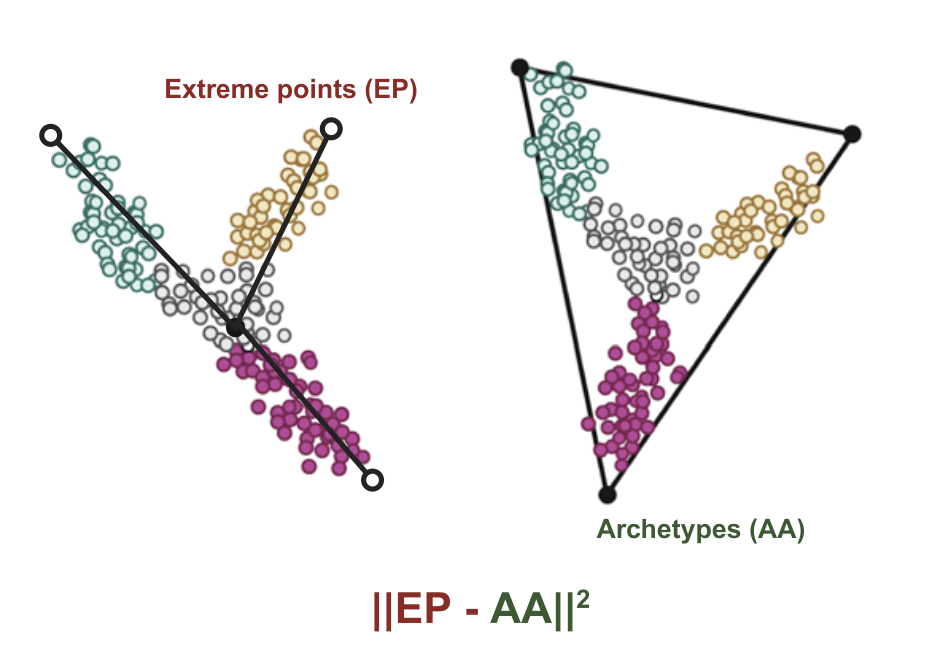
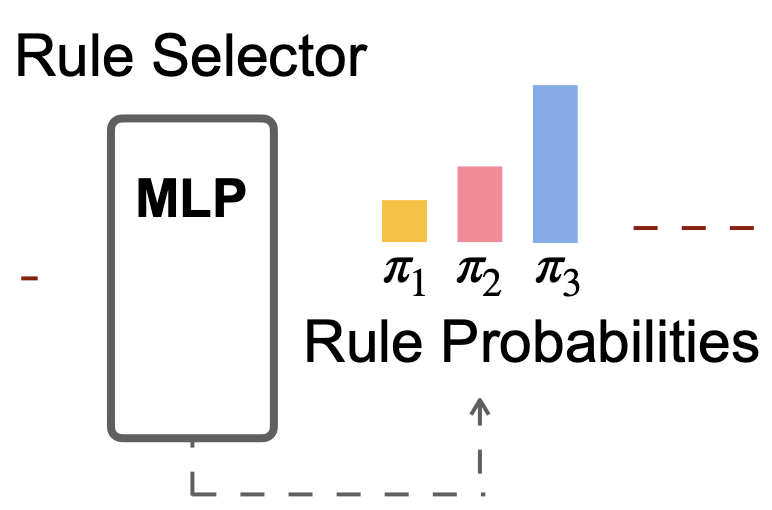
Predicting and controlling cancer evolution
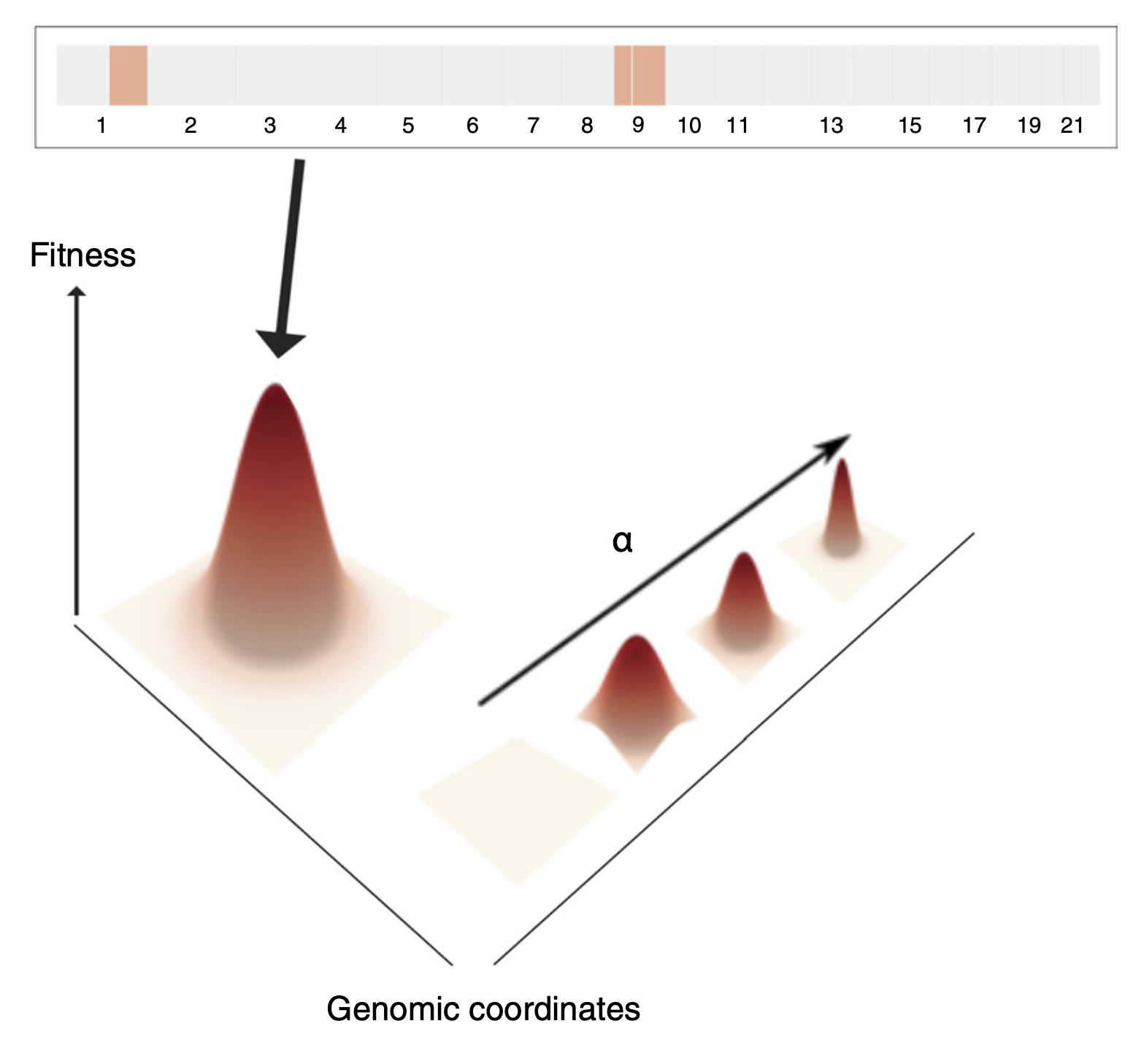
We have pioneered the use of evolutionary theory applied to cancer genomic data and designed amongst the first computational methods combining AI with mechanistic modelling for oncology. Our lab focuses on deciphering the dynamics of cancer growth, progression and treatment resistance using mathematical and computational approaches applied to multi-omic data, with the objective of predicting and controlling the disease. We also develop experimental evolution approaches to study cancer drug resistance, bringing the theory+experiment approach from physics to biology. We are part of the Computational Biology Research Centre at Human Technopole, a new life science institute in Milan, Italy.

Funded by