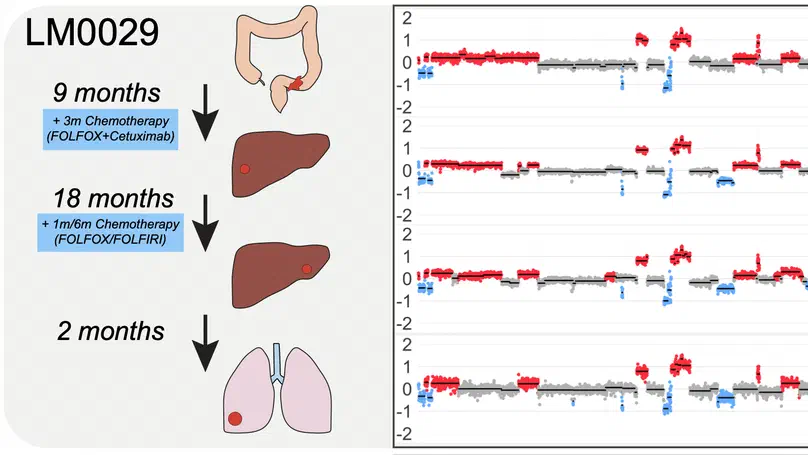
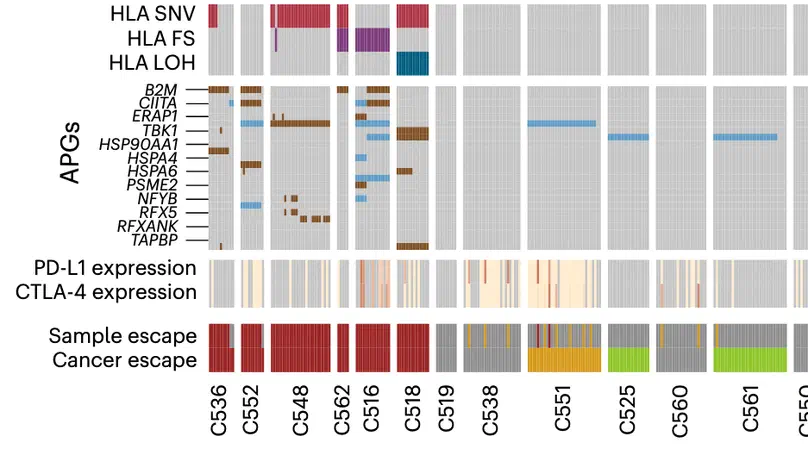
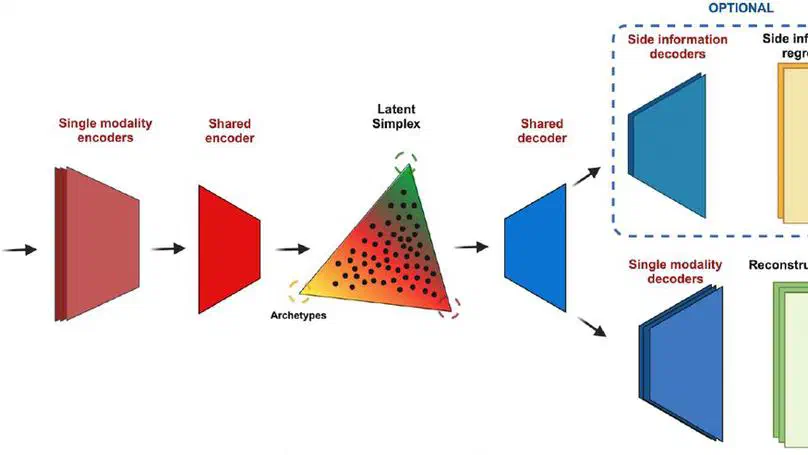
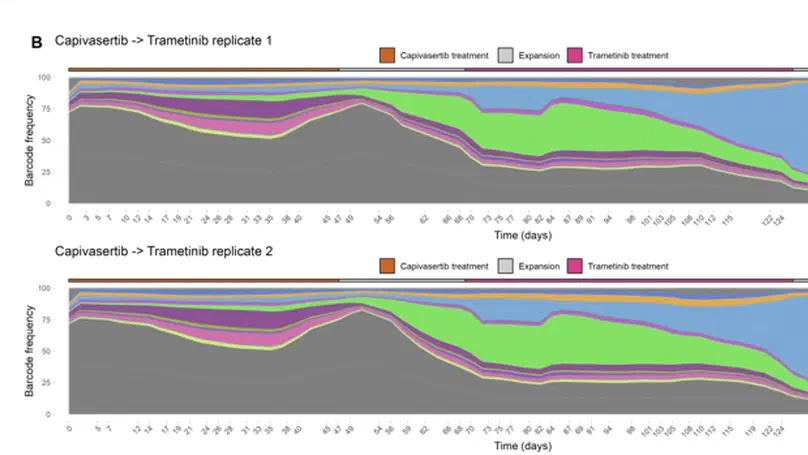
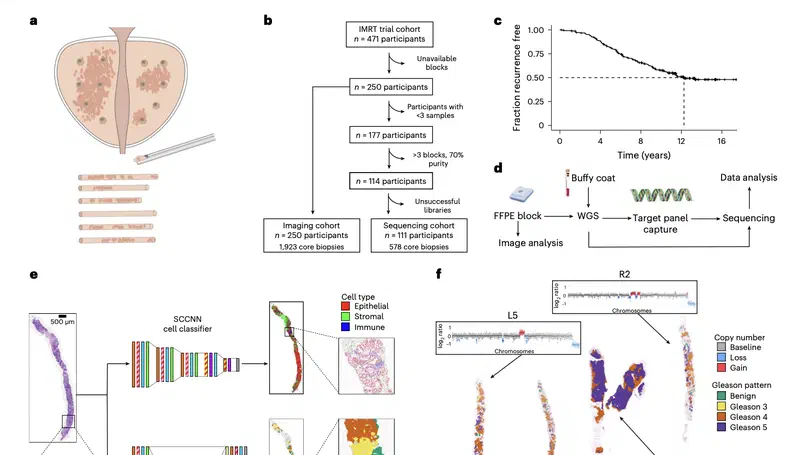
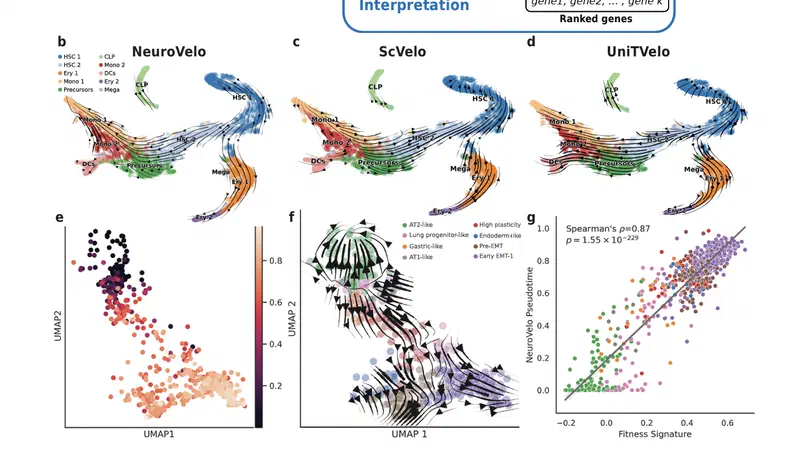
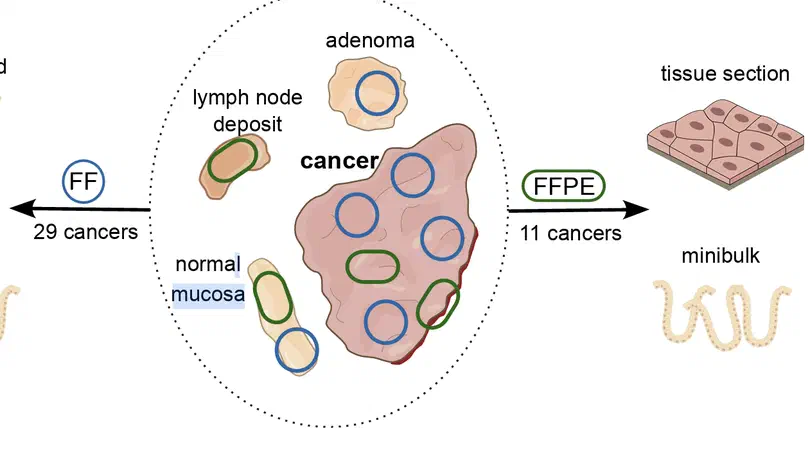
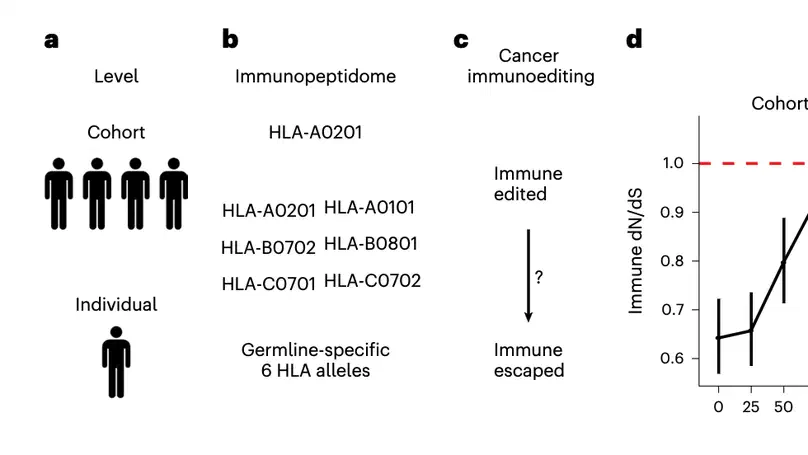
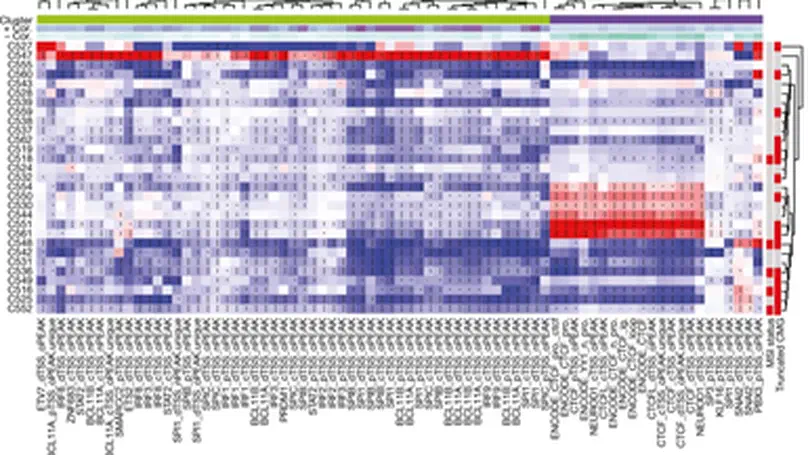
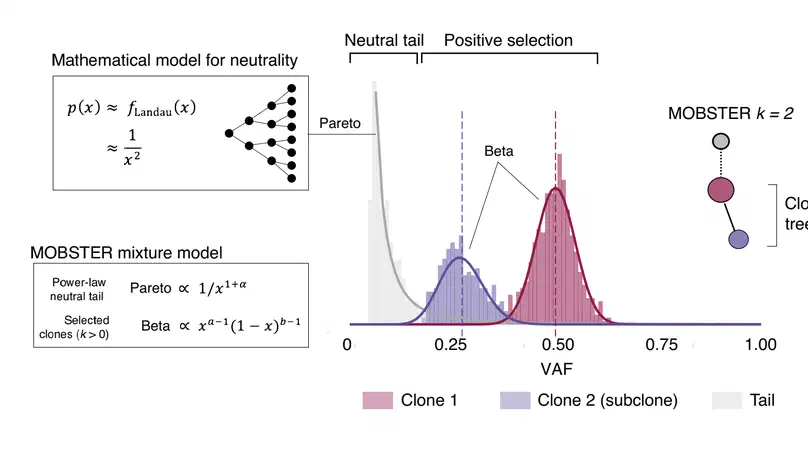
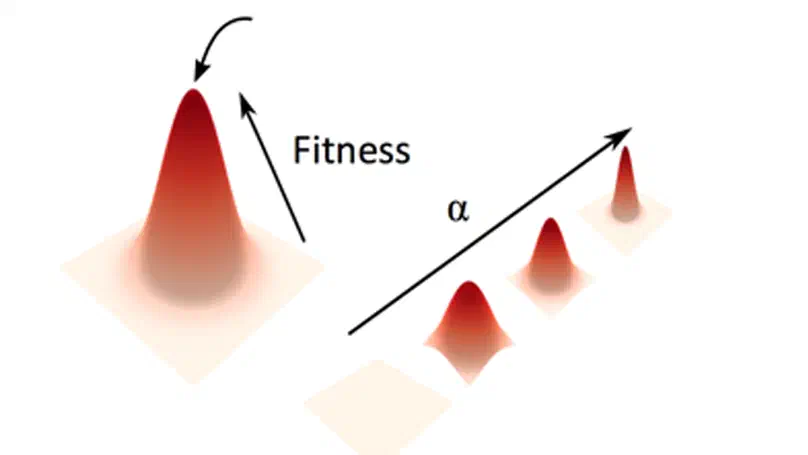
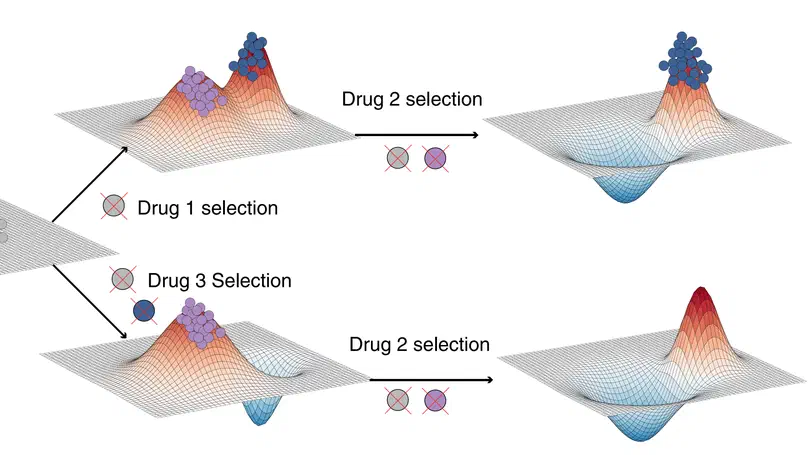
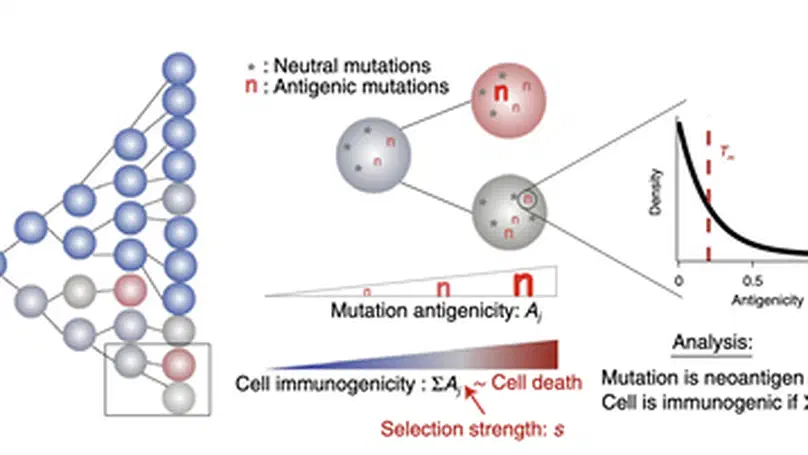
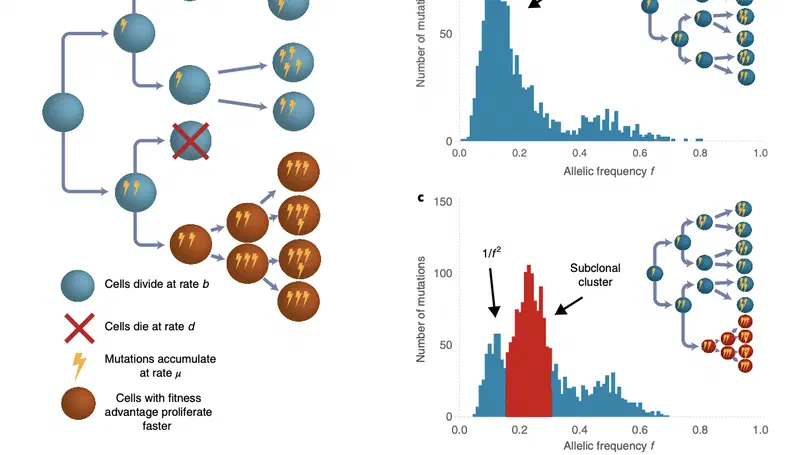
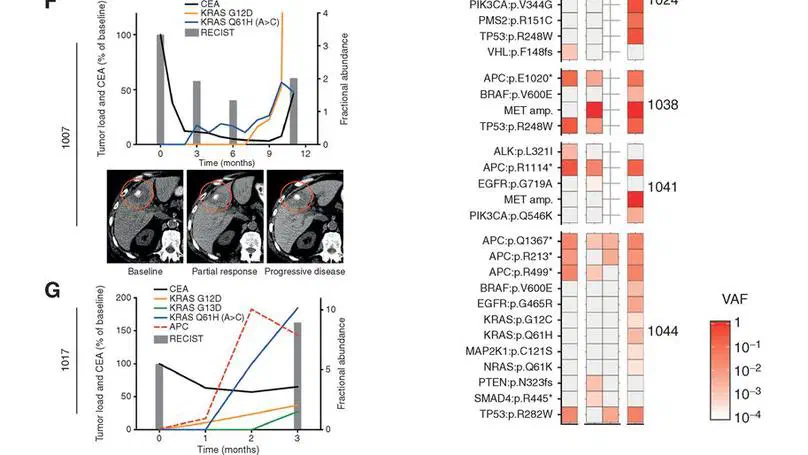
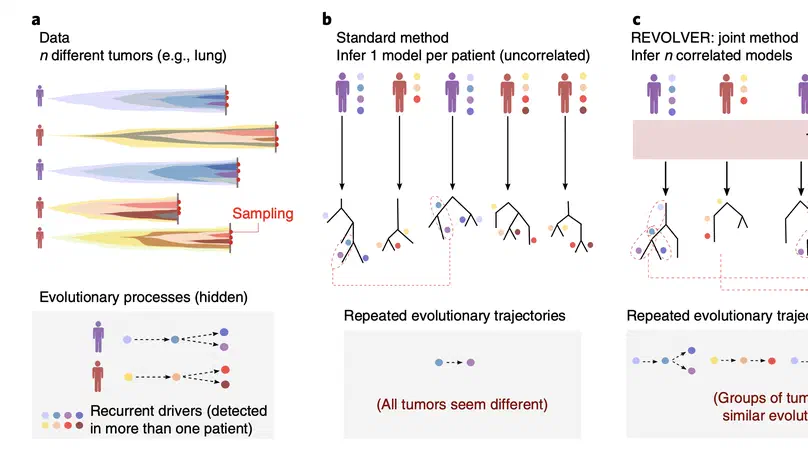
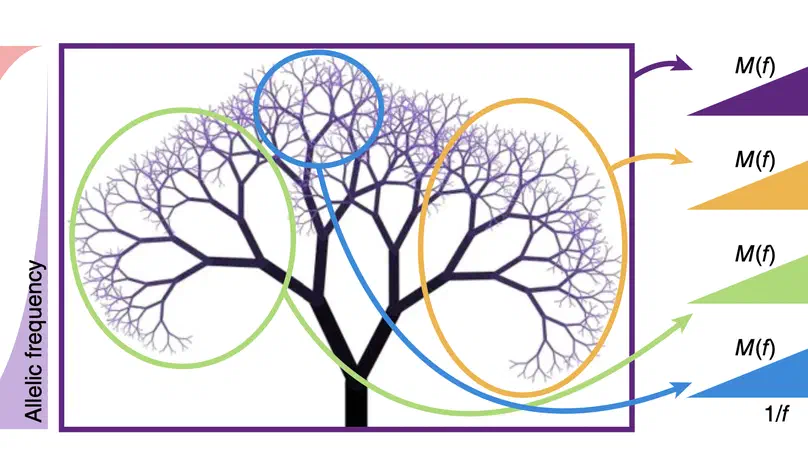
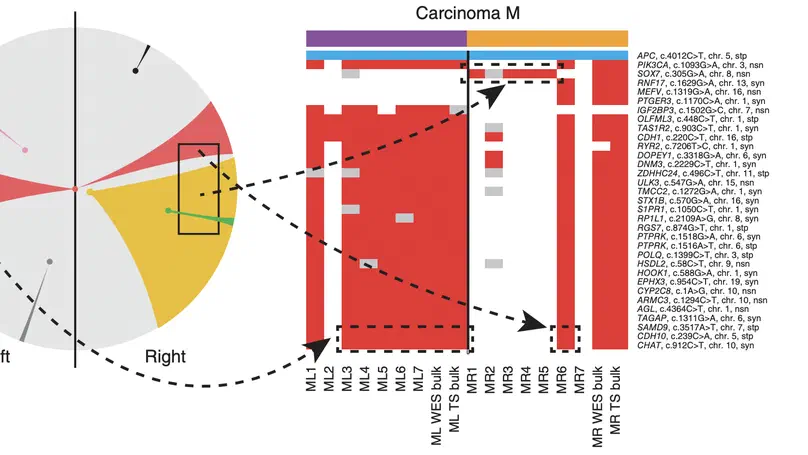
Aneuploidy, defined as the loss and gain of whole and part chromosomes, is a near-ubiquitous feature of cancer genomes, is prognostic, and likely an important determinant of cancer cell biology. In colorectal cancer (CRC), aneuploidy is found in virtually all tumours, including precursor adenomas. However, the temporal evolutionary dynamics that select for aneuploidy remain broadly uncharacterised. Here we perform genomic analysis of 755 samples from a total of 167 patients with colorectal-derived neoplastic lesions that cross-sectionally represent the distinct stages of tumour evolution, and longitudinally track individual tumours through metastasis and treatment. Precancer lesions (adenomas) exhibited low levels of aneuploidy but high intra-tumour heterogeneity, whereas cancers had high aneuploidy but low heterogeneity, indicating that progression is through a genetic bottleneck that suppresses diversity. Individual CRC glands from the same tumour have similar karyotypes, despite prior evidence of ongoing instability at the cell level. Pseudo-stable aneuploid genomes were observed in metastatic lesions sampled from liver and other organs, after chemo- or targeted therapies, and late recurrences detected many years after the diagnosis of a primary tumour. Modelling indicates that these data are consistent with the action of stabilising selection that ‘traps’ cancer cell genomes on a fitness peak defined by the specific pattern of aneuploidy. These data show that the initial progression of CRC requires the traversal of a rugged fitness landscape and subsequent genomic evolution, including metastatic dissemination and therapeutic resistance, is constrained by stabilising selection.